 Cambridge University Teaching Hospital
Cambridge University Teaching Hospital
Vitreoretinal Research
The Vitreoretinal Research Group at Cambridge University was established in 1993 to investigate and understand more clearly the causes, prevention and repair of retinal detachment.
In contrast to many other blinding retinal disorders, retinal detachment is both relatively common and potentially preventable if those at risk could be more accurately identified.
Retinal Detachment - Causes
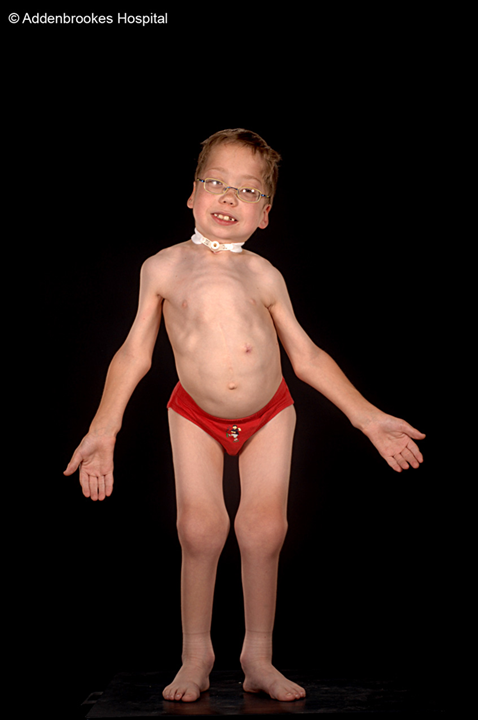
It is a relatively common misconception that retinal detachment occurs secondary to boxing or other direct trauma. In fact this is rarely the case. Our research over the last 27 years has been directed at investigating the genetic risk factors for retinal detachment, particularly in children where retinal detachment can be so devastating in terms of life-long visual loss. As a result of this research, in 2011 our service was commissioned by NHS England to provide the Highly Specialised diagnostic Service for Stickler syndrome – the commonest cause of retinal detachment in children and retinal detachment running in families. In 2016 our service was awarded the Bayer ophthalmology "gold" award for excellence in translation of research into patient care and receives referrals from all over the UK and the Republic of Ireland as well as further afield in Europe.
Retinal Detachment – Prevention
As a result of these recent advances in molecular genetic analysis, it is now possible to stratify with far greater accuracy the risk of retinal detachment, particularly those with an affected sibling or family history. By identifying those at highest risk we have developed a strategy for surgical prevention which hugely reduces the incidence retinal detachment.
Retinal Detachment – Repair
Our Vitreoretinal Service offers a 7 day a week, 365 day a year service for the emergency management of all aspects of retinal and vitreous disorders including retinal detachment, eye trauma, severe diabetic eye disease, all aspects of macular surgery (including macular hole, macular pucker) and retinal detachment repair in children. In addition, we receive referrals from other units for the management of the complications of cataract surgery and repair of recurrent retinal detachment for which primary surgical repair has been unsuccessful.
The use of liquid silicone for retinal detachment repair was pioneered in Cambridge by Mr JD Scott and we are currently collaborating with both the Department of Chemical Engineering and Biotechnology, University of Cambridge and the British Ophthalmic Surveillance Unit (BOSU) investigating further the biomechanics of the intraocular use of silicone and perfluorocarbon liquids for retinal surgery.
Allied speciality collaborations
We currently have ongoing research collaborations with the University of York, the Medical Research Council, the University of Birmingham NHS Foundation Trust and the Dept of Clinical Neurosciences, University of Cambridge investigating duel-sensory loss and markers of osteoarthritis .

Our Significant Research and Service milestones
- 1994 - Identification of genetic heterogeneity in Stickler syndrome
- 1996 - Identification of the gene for Type 2 Stickler syndrome
- 2000 - Identification of the genetic basis for "ocular only" Stickler syndrome
- 2011 - NHS England commission Cambridge Vitreoretinal Service to provide the Highly Specialised national service for patients with Stickler syndrome
- 2013 - Identification of a novel recessive variant of Stickler associated with total congenital deafness
- 2014 - Surgical protocol published to reduce the risk of retinal detachment in Type 1 Stickler syndrome
- 2016 - Identification of genetic variant in the Stickler gene associated with retinal detachment in general population
Useful Links:
- https://www.brc.cam.ac.uk/principal-investigators/martin-snead/
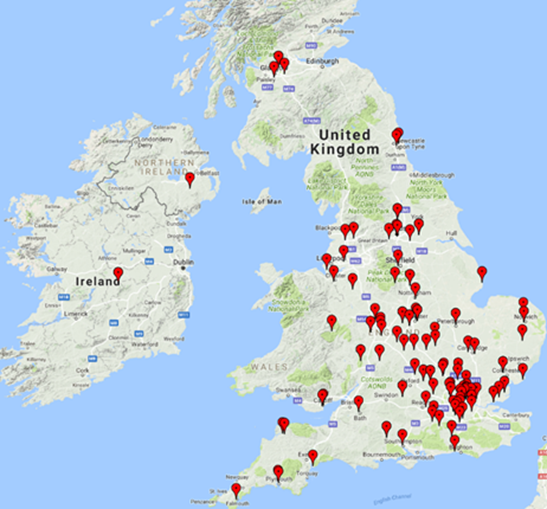
Our Vitreoretinal Service receives referrals from all over the UK and the Republic of Ireland as well as further afield in Europe.
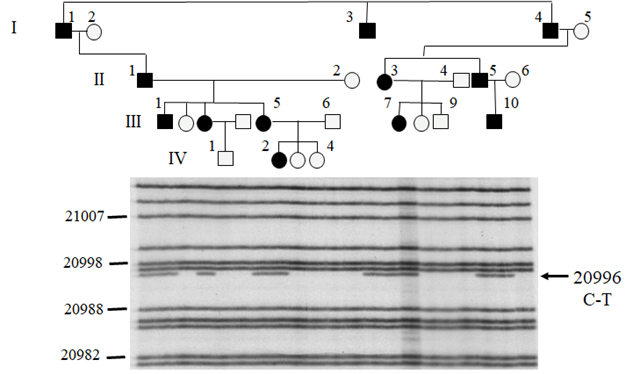
Genetic analysis identifies those family members at risk of retinal detachment (solid symbols)
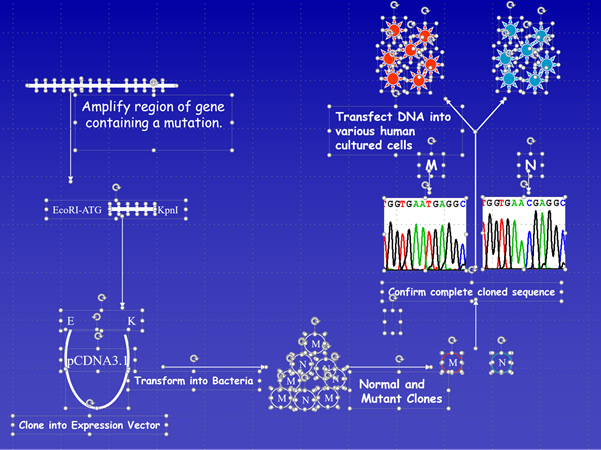
We have also developed a novel mini-gene laboratory model to determine the pathogenicity of deep intronic variants identified by whole gene sequencing affecting splicing. This functional mini-gene analysis is also commissioned as part of the NHS England specialist service.

MRC Epidemiology Unit Institute of Metabolic Science Cambridge Biomedical Campus.
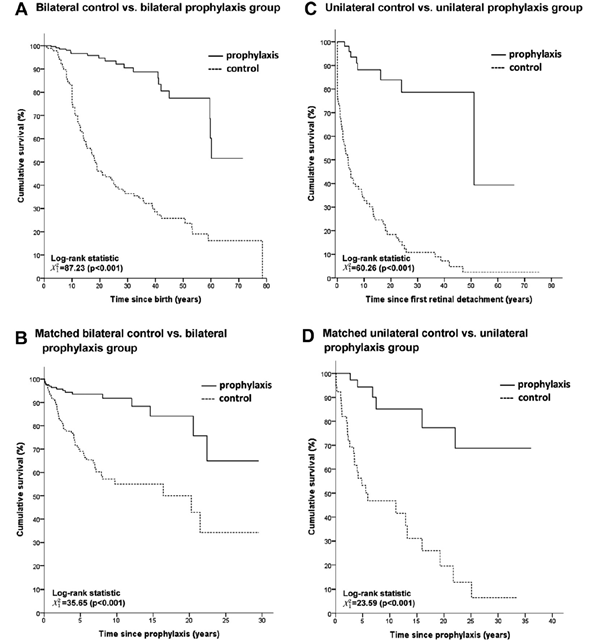
Kaplan-Meier survival curves for patients with and without prophylaxis against retinal detachment – Type 1 Stickler syndrome.
In association with School of Health and Related Research ScHARR (University of Sheffield) and MRC Epidemiology Unit, Institute of Metabolic Science, Cambridge Biomedical Campus
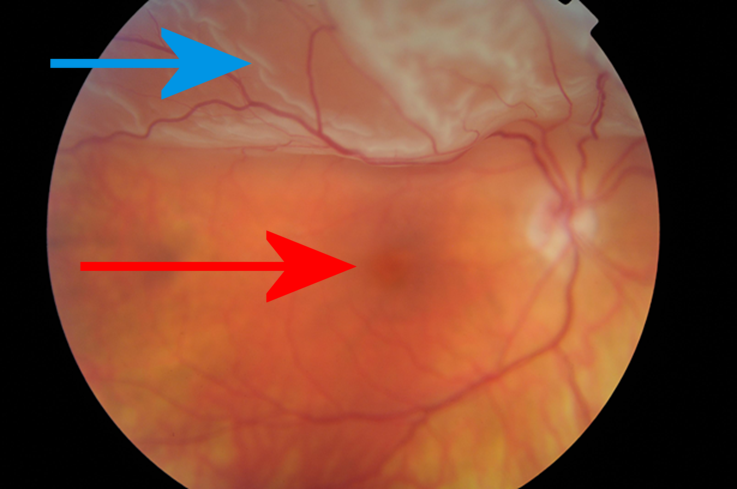
Retinal detachment (blue) rapidly progressing to threaten macular (central) vision. Urgent surgical repair is required to maintain good vision.

Our research includes a collaborative study with both the British Ophthalmological Surveillance Unit [BOSU] and the University of Cambridge Dept. of Chemical Engineering and Biotechnology investigating the use of intraocular liquid silicone and perflourocarbon liquids in the repair of retinal detachment.

|
Make a donation to the Vitreoretinal Research Group |